Bootstrap confidence intervals and p-values for evaluating the significance/stability of the block-weight vectors produced by S/RGCCA.
Arguments
- rgcca_res
A fitted RGCCA object (see
rgcca).- n_boot
The number of bootstrap samples (default: 100).
- n_cores
The number of cores used for parallelization.
- verbose
A logical value indicating if the progress of the bootstrap procedure is reported.
Value
A rgcca_bootstrap object that can be printed and plotted.
- n_boot
The number of bootstrap samples, returned for further use.
- rgcca
The RGCCA object fitted on the original data.
- bootstrap
A data.frame with the block weight vectors and loadings computed on each bootstrap sample.
- stats
A data.frame of statistics summarizing the bootstrap data.frame.
Examples
# Bootstrap confidence intervals and p-values for RGCCA
data(Russett)
blocks <- list(
agriculture = Russett[, seq(3)],
industry = Russett[, 4:5],
politic = Russett[, 6:8]
)
fit_rgcca <- rgcca(blocks, ncomp = 1)
boot_out <- rgcca_bootstrap(fit_rgcca, n_boot = 20, n_cores = 1,
verbose = TRUE)
print(boot_out)
#> RGCCA bootstrap object obtained with 20 bootstrap samples.
plot(boot_out, type = "weight", block = 1:3, comp = 1,
display_order = FALSE)
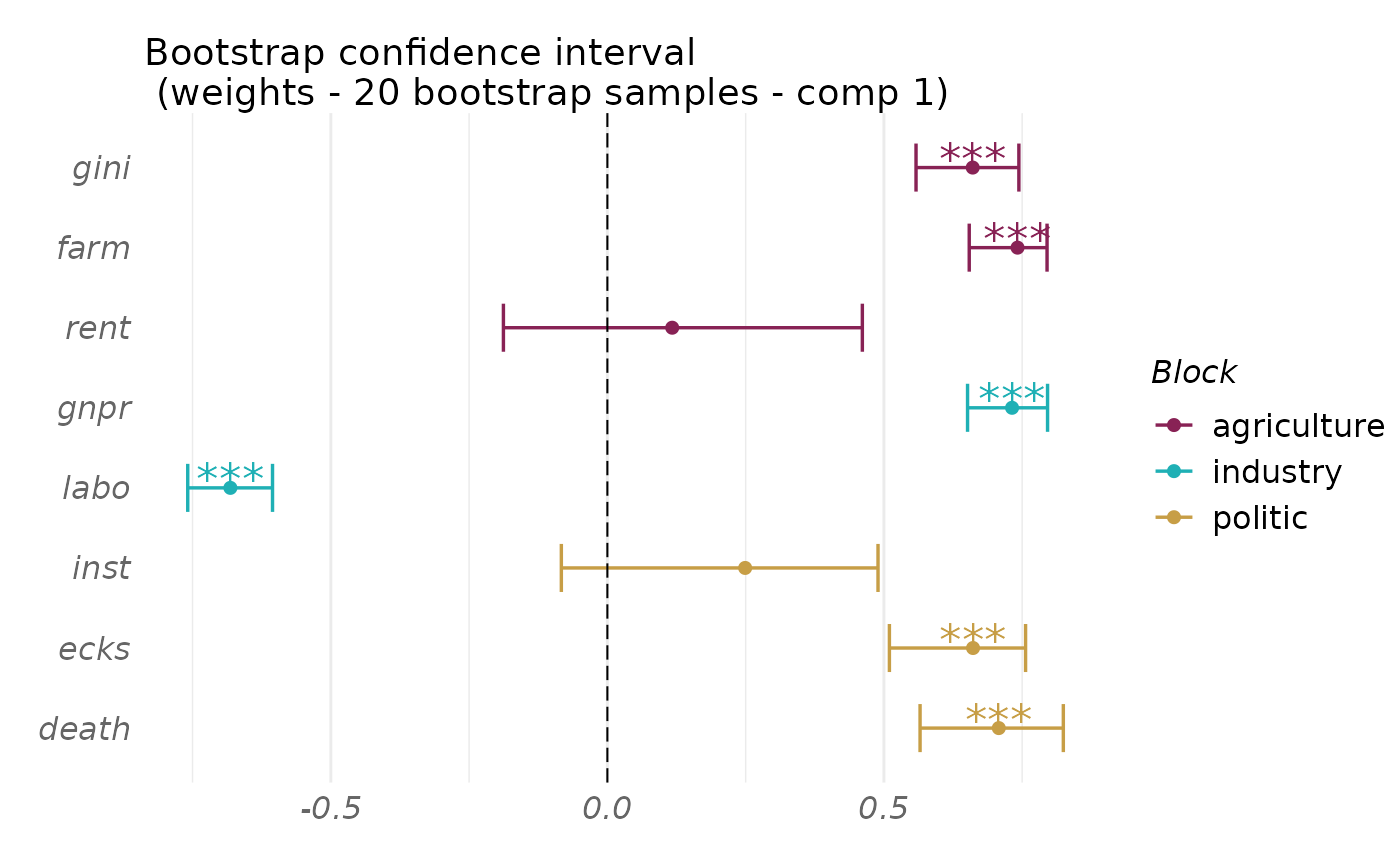 if (FALSE) { # \dontrun{
# Download the dataset's package at http://biodev.cea.fr/sgcca/ and install
# it from the package archive file.
# You can do it with the following R commands:
if (!("gliomaData" %in% rownames(installed.packages()))) {
destfile <- tempfile()
download.file(
"http://biodev.cea.fr/sgcca/gliomaData_0.4.tar.gz", destfile
)
install.packages(destfile, repos = NULL, type = "source")
}
data("ge_cgh_locIGR", package = "gliomaData")
blocks <- ge_cgh_locIGR$multiblocks
Loc <- factor(ge_cgh_locIGR$y)
levels(Loc) <- colnames(ge_cgh_locIGR$multiblocks$y)
blocks [[3]] <- Loc
fit_sgcca <- rgcca(blocks, response = 3,
sparsity = c(.071, .2, 1), ncomp = 1,
scheme = "factorial",
verbose = TRUE
)
print(fit_sgcca)
boot_out <- rgcca_bootstrap(fit_sgcca, n_boot = 50, n_cores = 2)
plot(boot_out, block = 1:2, type = "weight",
comp = 1, n_mark = 300000,
display_order = FALSE)
} # }
if (FALSE) { # \dontrun{
# Download the dataset's package at http://biodev.cea.fr/sgcca/ and install
# it from the package archive file.
# You can do it with the following R commands:
if (!("gliomaData" %in% rownames(installed.packages()))) {
destfile <- tempfile()
download.file(
"http://biodev.cea.fr/sgcca/gliomaData_0.4.tar.gz", destfile
)
install.packages(destfile, repos = NULL, type = "source")
}
data("ge_cgh_locIGR", package = "gliomaData")
blocks <- ge_cgh_locIGR$multiblocks
Loc <- factor(ge_cgh_locIGR$y)
levels(Loc) <- colnames(ge_cgh_locIGR$multiblocks$y)
blocks [[3]] <- Loc
fit_sgcca <- rgcca(blocks, response = 3,
sparsity = c(.071, .2, 1), ncomp = 1,
scheme = "factorial",
verbose = TRUE
)
print(fit_sgcca)
boot_out <- rgcca_bootstrap(fit_sgcca, n_boot = 50, n_cores = 2)
plot(boot_out, block = 1:2, type = "weight",
comp = 1, n_mark = 300000,
display_order = FALSE)
} # }